Product Calculations
1. In the main Cerius2 window change form stick view to ball and stick view by left clicking on the light brown box labeled stick directly below the drop down menu bar.
2. In the drop down menu left click on build select 3D-sketcher and the sketcher window will open and look like this:
3. In the sketcher window left click on the sketch button that looks like this:
.
4. Be sure that directly to the right of the sketch button the carbon atom is selected, indicating that you will be drawing a carbon atom.
5. Draw a carbon atom by left clicking in the Cerius2 Models window once and then moving the pointer outside the model window. If you happen to make a mistake left click on the delete atom button
and left click in the model window on the atom that you wish to delete.
6. Next to the sketch button in the sketch window left click on the drop down button
next to the blue box where there should presently be a C and change to a N indicating that you will be drawing a nitrogen atom next.
7. Left click in the model window and then remove the pointer once again.
8. Draw a hydrogen atom in the model window using the same technique.
9. To bond these atoms select the edit bond button
in the sketch window and to the right of the edit bond button left click and hold on the light brown button labeled single. A drop down menu should appear containing all available bond types. Select single bond and move to the model window and left click on the H then on the C. You should have drawn a single bond between them.
10. Now change to a triple bond and draw the bond between the carbon and the nitrogen.
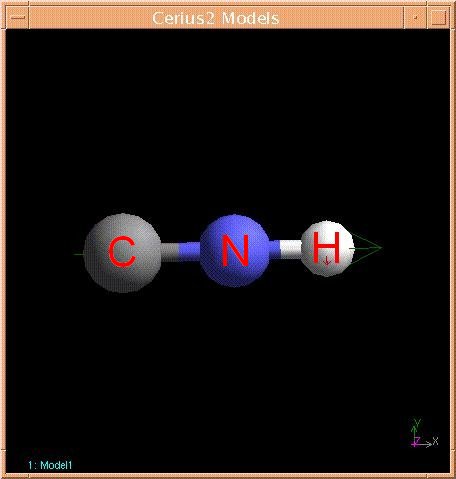
11. Left click and hold on the clean button in the lower left of the sketch window until the CNH molecule stops moving, indicating that the program has roughly minimized the molecules energy. Your CNH molecule should look something like this:
Possibly with a slightly longer CN bond since the clean button does not do a very good job of minimizing the energy of the molecule.
12. In the right portion of the visualizer window left click and hold on the light brown bar that is labeled BUILDERS1 and select from the menu that appears QUANTUM1.
13. In the tiled windows below left click on the MOPAC tab, sending it to the front. The visualizer window should now look like this:
14. Now left click on Run in the blue MOPAC menus, another window labeled MOPAC Run window will pop up. This is where you will be able to tailor the calculations that you will be doing on these molecules. The window will look like this, here the file name was changed, the calculation function was changed to geometry optimization and frequency, and the calculation method was changed to PM3. You will learn how to do that in the next steps.
15. First we will change the file name that this run will be saved under by left clicking in the blue box below the run button in the upper left hand corner, presently the name of the file should be mopac.
16. Change the file name to a suitable file name for the reactant calculations (i.e., cnh_reactant, do not use spaces in the file name) and hit enter.
17. Change the calculation task to geometry optimization and frequency calculation.
18. Left click and hold on the light brown button below the calculation function chooser and select the PM3 calculation method.
19. Further down in the MOPAC Run window you will find the electron spin, this specifies how many unpaired electrons are present in the molecule. For our CNH molecule there are no unpaired electrons so leave the spin as zero.
20. After all of the options you want are selected left click on the blue RUN button in the upper left had corner of the Run window. A small pop up window should appear as the computer calculates.
21. When the run window disappears the calculation is done. A graph window will pop up that contains the infrared spectra of the molecule. In the visualizer window in the right hand portion left click and hold on the analyze button. The properties that you may analyze are displayed here.
22. Chose files, the file analyze window will appear with the heat of formation and dipole moment shown in the lower portion. Left click on the Examine File button to show a text form of the output of the calculation. Scroll through the data and record the heat of formation, the enthalpy, the heat capacity, and the entropy all at 300K.
23. Left click and hold on the analyze button and select the vibrations.
24. With the model window visible select a frequency and left click the green animate button below the frequencies to animate the vibrations. To stop the vibration animation just left click on another frequency. These are the bends, stretches, and rotations that the molecule would be doing in its natural state.
25. Go ahead and close the vibrations window.
26. Now there are some very interesting molecular orbitals that we can view, but first for clarity we are going to change a viewing parameter so we can see the atoms a bit more clearly within the molecular orbitals. Left click on surfaces under the analyze section of the blue MOPAC menus, there is a transparency % selection in the lower left corner or the surfaces window. Type 50 in the blue box and hit enter, then close the window. Then left click and hold on analyze again and select orbitals. Another pop up window should appear showing the different orbitals and their corresponding energies.
27. Left click on the first orbital and then left click on the calculate button in the upper left hand corner of the orbitals window. Another window will pop up and show that the orbital is being calculated. After the calculation is finished the CNH molecule in the models window should be surrounded by a somewhat spherical light blue orbital.
28. Left click on each orbital and calculate each to see what they look like, there are some really cool looking ones, then go ahead and close the orbitals window.
Now lets move on to the calculations.