
| Back to Main Page | Professor Vincent L. Pecoraro | Group Information | Publications | Lectures and Conferences |
| Metallopeptide Project | Vanadium Project | Metallacrown Project | Gentamicin Project |
| Inorganic Chemistry | Related Links |

The oxygen evolving complex, OEC, is part of photosystem II, PSII. This enzyme catalyzes the oxidation of two molecules of water to yield one molecule of dioxygen, four protons, and four equivalents of electrons, as shown in Scheme 1 below.
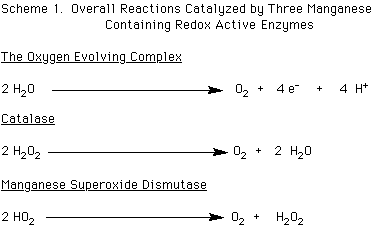
The OEC is part of the overall photosynthetic system that harnesses light energy for the eventual reduction of carbon dioxide and water to form sugars in green plants and algae. The core of the OEC contains a cluster of four manganese ions. The OEC is successively oxidized in four one electron steps to yield four equivalents of electrons that are used as reducing equivalents. Most, if not all, of these oxidations occur on the tetranuclear core of manganese ions. After the fourth oxidation, a molecule of dioxygen is released, and the OEC returns to a lower overall oxidation state. This cycle was established by studying the periodic bursts of dioxygen that occurred with every fourth flash of light given to preparations of photosynthetic centers by Kok in the early 1970's, and has been named the S-state cycle, which proceeds from S0 to S4. Each electron drawn from the OEC is transferred via a redox active tyrosine residue, know as tyrosine Z, to an oxidized chlorophyll molecule that was excited and oxidized by absorbed light energy. The electron that had been lost from the chlorophyll then goes on to be utilized as a reducing equivalent further along the line in the photosynthetic system. (Debus, R. J., Biophys. et. Biochim. Acta, 1992, 1102, 269; Oxygenic Photosynthesis: The Light Reactions, Ort, D. R. and Yocum, C. F., Eds.; Kluwer Academic Publishers: 1996)
Catalase contains two manganese ions at its active site, while manganese superoxide dismutase, MnSOD, is a mononuclear enzyme. Both of these enzymes play a role in protecting an organism from the potential deleterious effects of the reduced forms of dioxygen, hydrogen peroxide and superoxide, respectively. While catalase to date has been found predominately in bacteria, manganese superoxide dismutase is more widespread, being found in a range of organisms from bacteria to the mitochondria of humans. Catalase catalyzes the disproportionation of two hydrogen peroxide molecules to two molecules of water and one of dioxygen. MnSOD catalyzes the dismutation of two molecules of superoxide of one molecule of hydrogen peroxide and one of dioxygen. The catalase enzyme completes its catalytic cycle in two two-electron reduction and oxidation steps, while MnSOD engages in a one electron process. The overall processes are depicted in Scheme 1 above. (Pecoraro, V. L. et al. Chem. Rev., 1994, 94, 807; Manganese Redox Enzymes, Pecoraro, V. L., Ed.; VCH Publishers, New York: 1992).
One class of molecules that we have explored are based on the ligand H2salpn, N,N'-bis(salicylidene)-1,3-diaminopropane. With this ligand and derivatives thereof, we have prepared a family of manganese dimers in the Mn(IV)Mn(IV) oxidation state with two bridging µ-O2-'s to yield Mn2O2 cores (Larson, E. J., and Pecoraro, V. L. J. Am. Chem. Soc., 1991, 112, 3810). A crystal structure of the underivatized dimer, [Mn(IV)(salpn)( µ-O)]2 appears in the link to this page, and is also shown in figure 1 below.
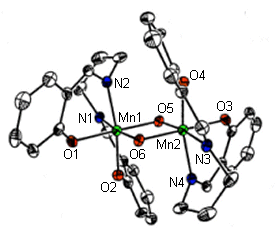
Figure 1. Crystal structure of [Mn(IV)(salpn)(O)]2
This [Mn(IV)(salpn)( µ-O)]2 complex structurally resembles a part of the OEC Mn-assembly, as it has a Mn-Mn separation of 2.7 , precisely the distance deduced for the Mn-Mn separation in the lower S-states of the enzyme. This dimer has proven particularly useful for our studies of manganese chemistry related to the function of the OEC. In 1991, our group reported that this complex is a very competent catalase mimic, and catalytically disproportionates hydrogen peroxide (Larson, E. J., and Pecoraro, V. L. J. Am. Chem. Soc., 1991, 112, 7809). This is interesting in light of the possible centered catalase-like reaction of the OEC. Another key aspect of this system has been our ability to protonate the oxo-bridges of the compound, and consequently to probe changes in the physical properties of this system. Our data has shown that a broad range of pKa's for the protonation of the oxo-bridges exists for this family of complexes and that there is also a broad range of reduction potentials, which indicates how nature may easily be able to control the potential at the tetranuclear manganese cluster of the OEC (Baldwin, M. J., et al., Photosynth. Research, 1993, 38, 303). Magnetic susceptibility studies have shown that successive protonation of the two oxo-bridges reduces the coupling of the two manganese centers to one another. Furthermore, these protonations lead to a lengthening of the Mn-Mn vector in the complex (Baldwin, M. J., et al., J. Am. Chem. Soc., 1994, 115, 11349).
These results may have implications for the functioning of the OEC. New proposals regarding the oxidation of water to dioxygen now suggest that the successive oxidations of the OEC may occur via a hydrogen atom abstraction process, thus coupling the removal of an electron with a concurrent loss of a proton, possibly from a water bound to one of the manganese ions in the OEC. Based on recent studies of tyrosine Z, it has been suggest that the H-atom abstraction is conducted by tryosine Z, the electron transfer agent between the OEC and the chlorophyll. We have utilized our system of [Mn(IV)(salpn)( µ-O)]2 complexes to explore the feasibility of the H-atom abstraction proposal. To do that, we utilized the properties of this system to calculate the homolytic bond dissociation energy, HBDE, for the loss of an H-atom from the protonated oxo-bridges of these complexes. Our data suggest that such a process is feasible (Baldwin, M. J. and Pecoraro, V. L. J. Am. Chem. Soc., 1996, 118, 11325-11326).
A second class of dimeric compounds that has provided many exciting results is based on the ligand H3(2-OHsalpn), N,N'-bis(salicylidene)-2-hydroxy-1,3-diaminopropane. This system is unique in many ways and has allowed us to explore the chemistry of the catalase enzyme (Gelasco, A., et al. Inorg. Chem., 1997, 36, 1829). The general structure of this complexes is the ligand binding to both manganese ions, with µ-O- alkoxide moieties from the ligand bridging the two manganese ions. The general structure of this family of complexes is shown by the crystal structure in Figure 2.
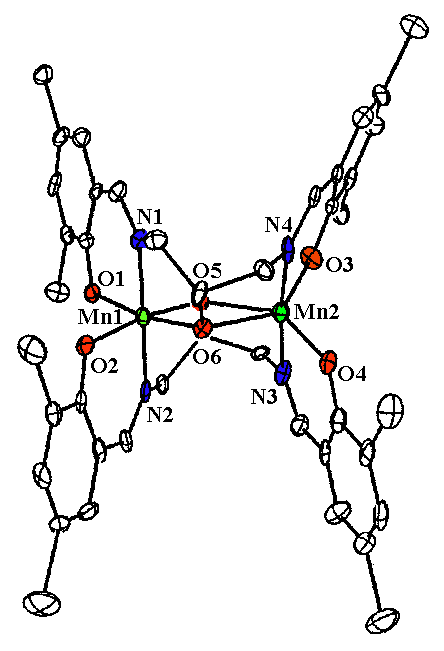
Figure 2. X-ray crystal structure of [Mn(III)(2-OHsalpn)]2. The ligand in this complex was prepared with 5-nitrosalicylaldehyde.
One of the key features of this system is that it is allows one to produce a series of complexes in four different overall oxidation states with only minor structural changes. These dimers have been prepared in the Mn(II)Mn(II), II/III, III/III, and III/IV oxidation states, a rare example of such a range of oxidation states with one ligand. This is key, since the catalase enzyme also exhibits this same set of oxidation states in its as isolated form. The catalase is considered to shuttle between the II/II and III/III oxidation states. When a one-electron process occurs to yield a II/III oxidation state, an ensuing two electron oxidation from the II/III state of the enzyme leads to the III/IV oxidation state, which is not active and only slowly over time is reduced back to a catalytically viable oxidation state. The [Mn(III)(2OHsalpn)]2 and [Mn(II)(2-OHsalpn)]2 complexes have been shown to be competentin the disproportionation of hydrogen peroxide, and that one is generated from the other if only turn over is allowed to occur. Thus the initial and final catalytic compounds have been established. Furthermore, this system exhibits saturation kinetic behavior. A catalytic cycle for this system is proposed in Scheme 2. (Gelasco, A., et al., J. Am . Chem. Soc., in press).
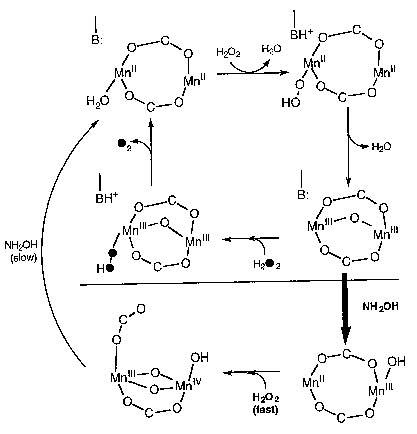
Scheme 2. A proposal for the catalytic cycle of Catalase based on the kinetics of hydrogen disproportionation of the two complexes.
While the reaction with hydrogen peroxide leads to disproportionation, the reaction with alkyl peroxides leads to the generation of free radicals. This reaction has been explored with t-butylhydroperoxide and the results of the reaction with cyclohexene indicated that this is a radical reaction. (Caudle, M. T., et. al. Inorg. Chem., 1996, 35, 3577).
Another unique feature of this system is its ability to bind an additional small molecule on one of the manganese ions, and to shift one of the bridging alkoxides into a terminal ligand status. Thus methanol and THF, for example, will bind to the Mn(III)Mn(III) complex, to form what is referred to as the unsymmetric dimer. This has been exploited to explore the binding of a hydroxide to the manganese and to probe such a complex spectroscopically. Again this may have implications for the OEC, and this family of complexes has been explored with respect to HBDE, as well (Caudle, M. T. and Pecoraro, V. L. J. Am. Chem. Soc., 1997, 119, 3415), since this dimer will bind a water molecule to form the unsymmetric dimer. The HBDE for the deprotonation of a water bound to manganese could then be obtained. Furthermore, in the case of hydroxide bound to the dimer, it was found that the presence of the hydroxide mediates the reduction potentials of the Mn(III)Mn(III) to the Mn(III)Mn(IV) dimer redox couple with respect to dimers without hydroxide bound, so that the Mn(III)Mn(IV) is more easily attained. Based on our newest results, and the H-atom abstraction hypothesis, we have completed a potential catalytic cycle of the OEC, which is presented in Scheme 3 (Pecoraro, V. L., et al. Pure and Applied Chemistry, Proceedings of the 8th International Conference on Bioinorganic Chemistry).
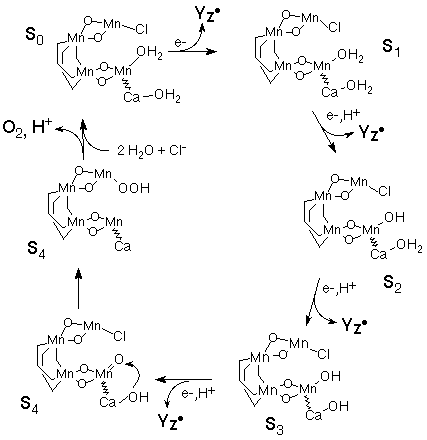
Scheme 3. A proposal for the S-state cycle of the OEC.
A new topic of research reapplies the H2salpn ligand in a new type of complex. It has been proposed that an imidazole bridge might exist in the OEC between two manganese ions. Our groups has been able to generate a dimeric complex bridged by an imidazole from the ligand 4,5-dicarboxyimidazole. A representative crystal structure is presented in Figure 3. Again we have been able to obtain this complex in several oxidation states. The Mn(IV)Mn(IV) oxidation state proved particularly unique, in that an unexpected EPR signal for a coupled Mn(IV)Mn(IV) system was observed (Caudle, M. T. et al. J. Am. Chem. Soc., 1997, 119, 9297).
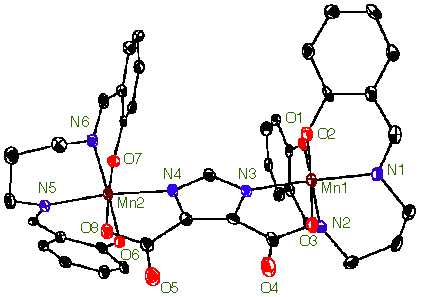
Figure 3. X-ray crystal structure of [(Mn(IV)(3,5-di-t-busalpn)) 2DCBI]. The H2salpn ligand in this complex is derivatized with t-butyl groups in the 3 and 5 positions on the phenyl rings. They have been omitted for clarity.
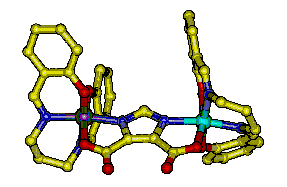
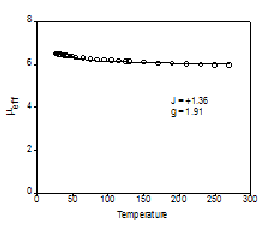 Significant attention has been focused on understanding the magnetic and
electronic structure of manganese dimers as these compounds have been used
extensively to model the spectroscopic signatures of the OEC. In all
cases examined for
[MnIIIMnIV(µ2-O)2]
3+
the magnetic coupling has been
antiferromagnetic and the ground state S=1/2. This has led to the
observation of the classic g=2 multiline feature in the epr spectra of
these complexes at cryogenic (4.2 K) temperatures. It has been speculated
for some time that g=2 multiline features should be observed for excited
states of ferromagnetically coupled
[MnIIIMnIV]7+ centers. In order to
test this hypothesis, we prepared
Mn2(III/IV)(dtsalpn)2DCBI shown in the
figure on the left. The incorporation of an imidazolate bridge causes a
net
ferromagnetic exchange coupling as shown in the plot of
µeff vs. T. The
coupling constant extracted from the fit of these data (J= + 1.4
cm-1)
confirms this assignment. Having an authentic ferromagnetically coupled
[MnIIIMnIV]7+ center, we obtained
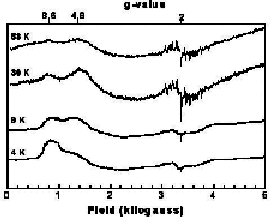
variable temperature X-band epr spectra
of the compound in solution. We observe that the dominant low temperature
feature is center at very low field at 4.2 K without discernable intensity
at g=2 for a multilane component. Upon warming the sample to 30 K, a new
g=2 feature develops which has the hallmark multilane feature. Thus, we
have demonstrated the long sought after excited state, g=2 multiline
signal. This work is published in J. Chem. Soc., ChemComm in 2003.
Significant attention has been focused on understanding the magnetic and
electronic structure of manganese dimers as these compounds have been used
extensively to model the spectroscopic signatures of the OEC. In all
cases examined for
[MnIIIMnIV(µ2-O)2]
3+
the magnetic coupling has been
antiferromagnetic and the ground state S=1/2. This has led to the
observation of the classic g=2 multiline feature in the epr spectra of
these complexes at cryogenic (4.2 K) temperatures. It has been speculated
for some time that g=2 multiline features should be observed for excited
states of ferromagnetically coupled
[MnIIIMnIV]7+ centers. In order to
test this hypothesis, we prepared
Mn2(III/IV)(dtsalpn)2DCBI shown in the
figure on the left. The incorporation of an imidazolate bridge causes a
net
ferromagnetic exchange coupling as shown in the plot of
µeff vs. T. The
coupling constant extracted from the fit of these data (J= + 1.4
cm-1)
confirms this assignment. Having an authentic ferromagnetically coupled
[MnIIIMnIV]7+ center, we obtained
variable temperature X-band epr spectra
of the compound in solution. We observe that the dominant low temperature
feature is center at very low field at 4.2 K without discernable intensity
at g=2 for a multilane component. Upon warming the sample to 30 K, a new
g=2 feature develops which has the hallmark multilane feature. Thus, we
have demonstrated the long sought after excited state, g=2 multiline
signal. This work is published in J. Chem. Soc., ChemComm in 2003.
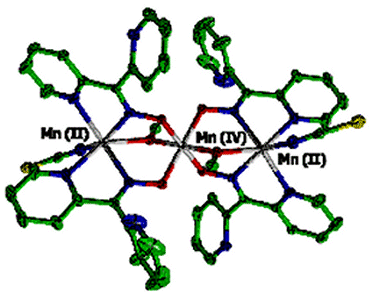 Arguably, second in importance to understanding the structure and
chemistry of the S4 state of the OEC, is the definition of
S0. This is
because S0 is the product state of the reaction process. Based
on XAS and
EPR studies, a MnIIMnIIIMnIV2
ensemble has been proposed for the
tetranuclear Mn cluster in S0. Such an oxidation state
formulation is rare
since it contains three different oxidation states. Under most
circumstances, MnII and MnIV would comproportionate
to make 2 MnIII.
However, something special about the OEC appears to stabilize the
different Mn oxidation states. We have prepared the first trinuclear Mn
complex containing MnII and MnIV. The compound is
Mn(II)Mn(IV)Mn(II)(pko)4(CH3O)
2(SCN)2.CH3OH
, which is synthesized by the
addition of 2,2'-dipyridyl etonoxime (Hpko) to a solution of sodium
hydroxide and NaSCN in CH3OH, with
MnCl2.4H2O. The neutral complex has 8
anions (2 thiocyanates, 2 µ-alkoxides and 4 pko- ligands),
and the Mn
oxidation state formulation of [Mn3]8+. There are
two possibilities for
the proper oxidation state assignment:
[MnIIMnIII2]8+ or
[MnII2MnIV]8+.
We can confirm that the later composition is correct by comparing the
properties of our new molecule to that of known trinuclear compounds of
composition [MnIIMnIII2]8+.
Key indicators supporting the proposed
oxidation state assignment are the structures of the central octahedral
MnIV ion and each MnII ion. The bond distances
around the Mn(IV) atom
average 1.91 E , and around each Mn(II) atom average 2.223 E. There is no
evidence of Jahn-Teller distortion, which is typical of Mn(III).
Arguably, second in importance to understanding the structure and
chemistry of the S4 state of the OEC, is the definition of
S0. This is
because S0 is the product state of the reaction process. Based
on XAS and
EPR studies, a MnIIMnIIIMnIV2
ensemble has been proposed for the
tetranuclear Mn cluster in S0. Such an oxidation state
formulation is rare
since it contains three different oxidation states. Under most
circumstances, MnII and MnIV would comproportionate
to make 2 MnIII.
However, something special about the OEC appears to stabilize the
different Mn oxidation states. We have prepared the first trinuclear Mn
complex containing MnII and MnIV. The compound is
Mn(II)Mn(IV)Mn(II)(pko)4(CH3O)
2(SCN)2.CH3OH
, which is synthesized by the
addition of 2,2'-dipyridyl etonoxime (Hpko) to a solution of sodium
hydroxide and NaSCN in CH3OH, with
MnCl2.4H2O. The neutral complex has 8
anions (2 thiocyanates, 2 µ-alkoxides and 4 pko- ligands),
and the Mn
oxidation state formulation of [Mn3]8+. There are
two possibilities for
the proper oxidation state assignment:
[MnIIMnIII2]8+ or
[MnII2MnIV]8+.
We can confirm that the later composition is correct by comparing the
properties of our new molecule to that of known trinuclear compounds of
composition [MnIIMnIII2]8+.
Key indicators supporting the proposed
oxidation state assignment are the structures of the central octahedral
MnIV ion and each MnII ion. The bond distances
around the Mn(IV) atom
average 1.91 E , and around each Mn(II) atom average 2.223 E. There is no
evidence of Jahn-Teller distortion, which is typical of Mn(III).
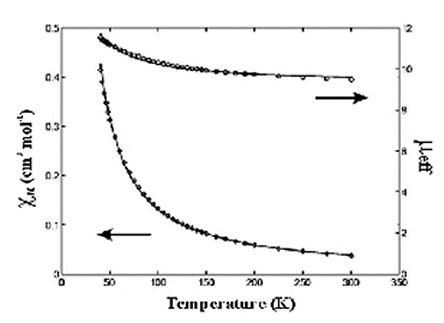
Interestingly, this compound shows
ferromagnetic exchange
interactions. The exchange parameter between the central MnIV
and terminal
MnII is J= +6.13 cm-1 and g=2.09 . The magnetic
ground state,
confirmed
by magnetization measurements at 4.5 K, is S=13/2 . The EPR spectral
features of this compound are also different from
[MnIIMnIII2]8+ trimers
, as MnIIMnIVMnII has unstructured, low
field transitions at 4.2 K while
MnIIIMnIIMnIII exhibit broad features,
with multiline character centered
around 1600 G. Most important, we have been able to assess whether X-ray
Absorption Spectroscopy can distinguish between the formulations
[MnIIMnIII2]8+ and
[MnII2MnIV]8+. The XANES
spectrum for
an authentic
Mn(III)2Mn(II) trimer, is somewhat narrower than that seen for
MnIIMnIVMnII or for its Cl-
analog. When the XANES spectra for
MnIIMnIVMnII were fit with a linear
combination of MnII, MnIII, and MnIV,
the best fits all had less than 15% MnIII, while the
corresponding fits
for MnIIIMnIIMnIII gave nearly 70%
MnIII and never gave composition of
MnIV > 15%. This work demonstrates that XANES should be able
to
discriminate the
MnIIMnIIIMnIV2
composition for the OEC.
In conclusion, we have been able to apply a broad range of model
complexes to broaden the understanding of the biological role of
manganese. We have produced a variety of information that has provided
new and exciting data for the field of bioinorganic manganese chemistry.
Our research in the areas described above is ongoing, with many
experiments left to be done. Thus, while we have accomplished much, there
are still numerous frontiers left for our laboratory to explore in the
context of the biological role of manganese.
| Back to Main Page | Professor Vincent L. Pecoraro | Group Information | Publications | Lectures and Conferences |
| Metallopeptide Project | Vanadium Project | Metallacrown Project | Gentamicin Project |
| Inorganic Chemistry | Related Links |