|

|
Biocatalysis
The direct, catalytic functionalization of C-H bonds presents a long withstanding challenge for synthetic chemists due, in part, to their
inert nature and ubiquity in organic compounds, making it difficult to achieve selective transformations. Transition-metal catalysts have been proven time and again
to be effective towards the activation of these bonds; however, their use presents a myriad of issues pertaining to the chemo-, regio-, and stereoselectivity in activating
a target C-H bond. Therefore, the usage of C-H functionalization strategies is often situationally-limited depending on the synthetic route. For example, directing groups are
commonly employed to dictate the position of the target C-H bond towards the metal center via coordination-assisted control. Though highly effective, this method can be limited,
requiring tactical design of the substrate. Within the many strategies available to activate C-H bonds, direct functionalization via metal-catalyzed group transfer reactions
is an attractive approach in organic synthesis to directly generate C-O, C-N, or C-C bonds.[1] Synthetic metalloporphyrin catalysts have been extensively studied for these reactions
due, in large part, from inspiration driven by the native C-H hydroxylation activity of heme-enzymes such as Cytochrome P450s.[2]
Carbene transfer reactions are of particular interest in the quickly developing field of organometallic biocatalysis. These reactions can produce new C-C, C-N, and C-S bonds,
and useful organic products such as cyclopropanes, amines and thioethers, among others. A number of transition metal complexes catalyze the decomposition of diazo reagents to form
an electrophilic metal-carbene unit that is then attacked by a nucleophilic reagent such as an olefin (for cyclopropanation) or an amine (for N-H insertion). In particular, a variety
of metalloporphyrin complexes including metals such as Fe, Co, Ru, Rh, and Ir have been effective towards catalyzing carbene transfer reactions.[3] These complexes catalyze thousands
of turnovers, and the second and third row transition metal complexes typically have long catalyst lifetimes, but also exhibit the aforementioned drawbacks of small molecule catalysts
where stereoselectivity is difficult to control and reactions are often performed in environmentally detrimental organic solvents. The Arnold and Fasan groups have demonstrated that
heme proteins such as Cytochrome P450BM3 and myoglobin (Mb) can be modified in order to produce very active and stereoselective carbene transfer catalysts.[4-6] Multiple rounds
of active-site mutagenesis and reactivity screening through directed evolution (invented by the Arnold group) can lead to improved product yield and stereoselectivity, though at
the expense of gaining a more fundamental understanding of how these modifications improve catalysis.[1] Rational design of a catalyst through active-site mutations and cofactor
modification is a more time-intensive approach, but can also lead to increased activity and selectivity of the system, and a better understanding of how the second coordination
sphere influences reactivity.
An alternative strategy to using the naturally occuring heme cofactor in biocatalysis is to incorporate non-natural hemes into heme proteins. Two strategies have evolved here, which are
the use of non-biological metals like Ru, Rh, Ir, etc.,[7,8] the use of non-natural heme derivatives (like porphycenes), and a combination of the two. We are interested in both of these
approaches, using Mb as the protein matrix of choice.
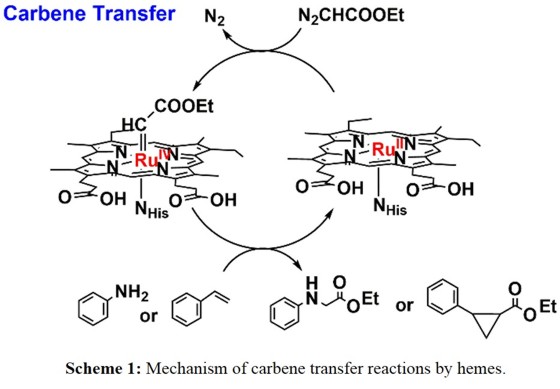
In particular, following the rational design approach, we designed Ru-modified Mb catalysts and studied their catalytic activity towards cyclopropanation and N-H insertion.
The mechanism for these transformations is shown in Scheme 1. For this purpose,
we have incorporated Ru mesoporphyrin IX (RuMpIX) into wild-type (wt) Mb and three His64 mutants (see Figure 1) and characterized these artificial enzymes using different spectroscopic methods.

For reactivity studies,
we used ethyl diazoacetate (EDA) as a carbene source, styrene derivatives for the cyclopropanation reactions and aniline derivatives for the N-H insertion reactions. These catalysts
exhibit moderate activity in the N-H insertion reaction of aniline derivatives (see Table 1), and low activity in the cyclopropanation of most of the styrene derivatives studied. RuH64DMb is able to achieve
the best diastereoselectivity, but exhibits low activity. Our results show that one has to be cautious when analyzing the outcomes of organometallic reactions in water: besides the reactivity
differences that result from electronic effects, solubility and acid/base equilibria can influence the outcome of reactions dramatically.

We also observed an interesting difference between
our RuMb catalysts and analogous iron systems studied in the literature, where application of Ru leads to significantly suppressed cyclopropanation reactivity. Our studies show that this is
not due to a lack of carbene transfer reactivity of RuMb, but to the contrary, that this is caused by the catalyst being too reactive, leading to degradation of the protein and the porphyrin,
and hence, fast catalyst deactivation (see Figure 2).
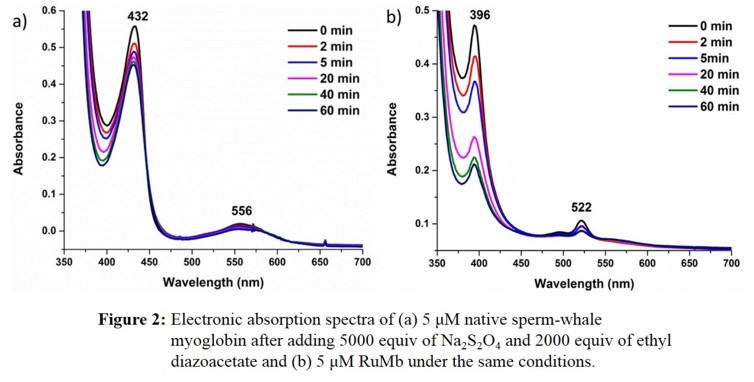
This is especially a factor when styrene substrates are used (for cyclopropanation) that have a low solubility in water. This result is encouraging as
it shows that RuMb has a great potential as a highly active carbene transfer catalyst, if the stability issues can be overcome. Our results also show that Mb mutants with hydrophobic
active site pockets show moderate increases in activity, at least in conjunction with the non-polar styrene and aniline derivatives studied here. Hence, even more active catalysts should result
from double or triple Mb mutants that further increase the hydrophobicity of the active site. To induce increased stereoselectivity in the cyclopropane products, bulky hydrophobic groups
should be used in these mutants. This is the next logical step in rational catalyst design that should lead to improved carbene transfer catalysts, and corresponding investigations are in progress.

In a complementary approach, we are interested in the usage of synthetic porphyrin derivatives in biocatalysis. As shown in Scheme 2, porphycene is a core isomer of porphyrins, leading to iron complexes
that are distinctively different from their heme analogs. In order to investigate whether the Fe-porphycene (FePc) complex is an active catalyst for carbene transfer reactions, we inserted FePc into the
apo-Mb matrix, generating "rMb" (reconstituted Mb), in collaboration with Prof. Takashi Hayashi's group at Osaka University. Carbene transter reactions with rMb using EDA and styrene (for cyclopropanation)
were then performed under anaerobic conditions in the presence of dithionite, and we found that FePc works as a good catalyst for the cyclopropanation of styrene in the myoglobin matrix. As shown in
Figure 3, rMb is a distinctively more active catalyst compared to native Mb ("nMb"). This increase in reactivity is rationalized with a much faster generation of the critical Fe-carbene intermediate (see Scheme 1)
in the rMb case compared to nMb. Theoretical investigations strongly support the fact that the strong ligand field of porphycene is useful for the efficient formation of the central carbene intermediate.
Further studies on the ability of rMb to catalyse carbene transfer reactions are underway.

References:
M. W. Wolf, D. A. Vargas, N. Lehnert
"Engineering of RuMb: Towards a Green Catalyst for Carbene Insertion Reactions"
Inorg. Chem. 2017, 56, 5623-5635
(ACS Select Virtual Issue: Engineered Biomolecular Catalysts)
K. Oohora, H. Meichin, L. Zhao, M. W. Wolf, A. Nakayama, J. Hasegawa, N. Lehnert, T. Hayashi
"Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species"
J. Am. Chem. Soc. 2017, 139, 17265-17268
B. W. Musselman, N. Lehnert
"Bridging and Axial Carbene Binding Modes in Cobalt Corrole Complexes: Effect on Carbene Transfer"
Chem. Commun. 2020, 56,14881-14884
(special issue: Bioinspired Metal Complexes for Chemical Transformations and Catalysis)
Literature:
[1] Che, C.-M.; Lo, V. K.-Y.; Zhou, C.-Y.; Huang, J.-S. Chem. Soc. Rev. 2011, 40, 1950-1975
[2] McQuarters, A. B.; Wolf, M. W.; Hunt, A. P.; Lehnert, N. Angew. Chem. Int. Ed. 2014, 53, 4750-4752
[3] Che, C. M.; Huang, J. S. Coord. Chem. Rev. 2002, 231, 151-164
[4] Coelho, P. S.; Brustad, E. M.; Kannan, A.; Arnold, F. H. Science 2013, 339, 307-310
[5] Coelho, P. S.; Wang, Z. J.; Ener, M. E.; Baril, S. A.; Kannan, A.; Arnold, F. H.; Brustad, E. M. Nat. Chem. Biol. 2013, 9, 485-487
[6] Bordeaux, M.; Tyagi, V.; Fasan, R. Angew. Chem. Int. Ed. 2015, 54, 1744-1748
[7] Key, H. M.; Dydio, P.; Clark, D. S.; Hartwig, J. F. Nature 2016, 534, 534-537
[8] Natoli, S. N.; Hartwig, J. F. Acc. Chem. Res. 2019, 52, 326�335
|
