|

|
Solar Energy Conversion into Chemical Fuels
Devices capable of generating dihydrogen from a renewable energy source are crucial for meeting the global dihydrogen consumption
demand in a sustainable way and for efforts to replace carbon-based fuel technologies with more sustainable alternatives.[1-3] Large-scale dihydrogen production
from protons and electrons, whether directly coupled to water oxidation photoanodes or to electolyzers powered by renewable energy sources, must inevitably be
facilitated by heterogeneous catalysts to allow for application of the catalysts in large-scale flow reactors.[4-5] To this end, electrocatalysts for dihydrogen
production must be designed that meet the demands of these systems, and, at the same time, are inexpensive, easy to assemble, and that can be prepared on a large
scale without difficulty. These electrocatalysts could either be built from catalytically active electrode materials, or from molecular catalysts immobilized
on the electrode surfaces.[6-8] The latter approach has the advantage that the molecular catalysts can be improved in a systematic manner and derivatized to meet
given application needs, as shown below. Our work is focused on the cathodic side of water splitting, i.e. the design of an intergrated device where a light-absorbing
semiconductor like GaP is coupled to a molecular proton reduction catalyst. Scheme 1 shows a schematic drawing of our target system.
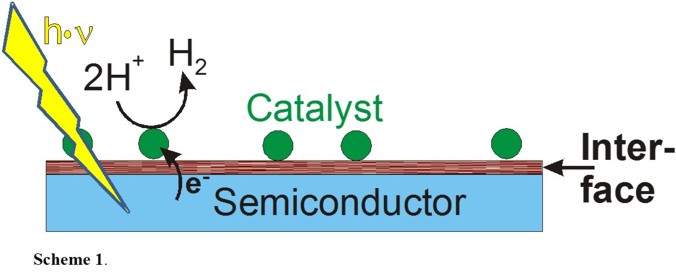
A substantial challenge in this regard is to design suitable interfaces that allow for the easy, yet stable and affordable, attachment of the catalysts to the electrode surfaces.
In this regard, we have demonstrated that reduced graphene oxide films can serve as inexpensive, yet extremely versatile interfaces to adsorb a variety of
Co-bis(benzenedithiolate),
[CoIII(bdt)2]2-, dihydrogen production catalysts to semiconductor electrode surfaces, yielding robust and highly active platforms for
electrocatalytic H2 generation. To test the applicability of these catalysts in the surface-immobilized state, we first studied simple test systems
where a thin film of reduced graphene oxide (RGO) is deposited on a fluorinated thin oxide (FTO) electrode, as shown in Scheme 2.
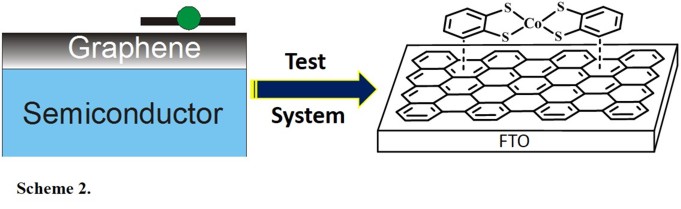
Simple dipping of these electrodes into a solution of a Co-bis(benzenedithiolate) complex is then sufficient to adsorp the catalyst on the electrode surface.
Not only do Co-bis(benzenedithiolate) complexes π-stack onto these "FTO/RGO" electrodes, they can also be adsorbed on cheap graphite electrodes, graphite powder,
and highly-ordered pyrolytic graphite (HOPG).
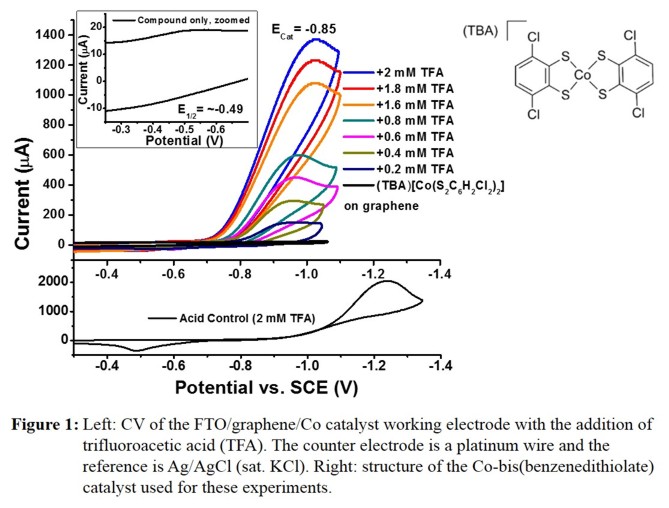
Figure 1 shows electrochemical data for the FTO/RGO/(TBA)[Co(S2C6H2Cl2)2] system as example, using the catalyst with
the 3,6-dichlorobenzenedithiolate ligand. Elecrochemical analysis by cyclic voltammetry (CV) shows a redox feature at -0.49 V (versus SCE), which corresponds to the
CoIII/CoII couple of the surface-immobilized catalyst. This feature persist upon extended cycling, indicating the stability of the adsorbed species
on the electrode surface. Addition of trifluoroacetic acid to the aqueous solution results in an increase in the cathodic current coinciding with disappearance of the anodic wave,
indicative of proton reduction at the electrode surface. Figure 2 illustrates the observed, linear dependence of the catalytic current on the acid concentration.
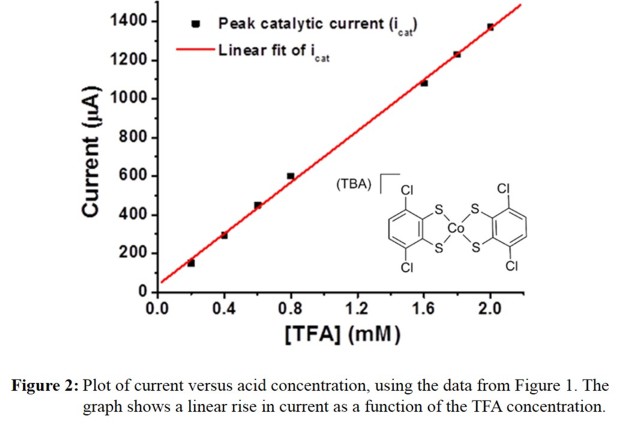
Further analysis of CV data shows turnover frequencies (TOF) in the range of 200 - 300 s-1 for this catalyst, depending on the type of graphitic
support used in the experiments. Figure 3 shows a comparison of different Co-bis(benzenedithiolate) derivates that we have studied.
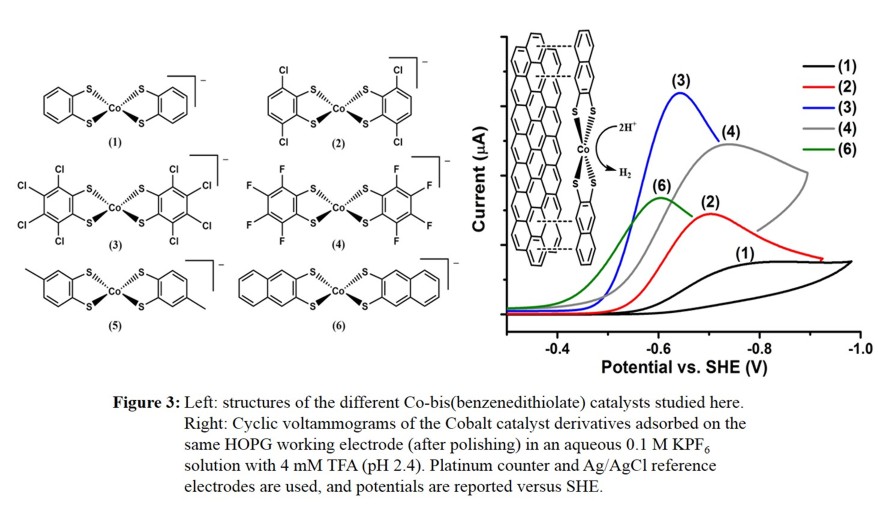
In summary, our surface-adsorbed Co-bis(benzenedithiolate) complexes exhibit excellent catalytic performance for dihydrogen production in moderately acidic aqueous solutions.
These catalysts are simple and inexpensive to prepare, air stable, and can easily be adsorbed onto graphitic surfaces using a small amount of catalyst material.
Catalyst loading increases as more electron withdrawing substituents are added to the ligand, though an even stronger effect is seen as the size of the aromatic ring system increases.
Overpotential decreases with increasing halide substitution of the benzenedithiolate ligand, although differences observed between Cl- and F-substituents are small.
Activity trends are evident across the
ligand series as turnover frequencies generally decrease with increasing halide substitution compared to the unsubstituted compound. Under bulk-electrolysis conditions (on HOPG),
halide-substituted derivatives slowly decrease in activity over the course of ~8 hours.
An average TOF of up to 320 s-1 over an 8-hour period is observed for the dichloro-substituted catalyst. Future work is now focused on (a) developing catalysts with
further improved surface adsorption (under turnover), and (b) immobilization of the catalysts on GaP.
References:
S. C. Eady, S. L. Peczonczyk, S. Maldonado, N. Lehnert
"Facile Heterogenization of a Cobalt Catalyst via Graphene Adsorption: Robust and Versatile Dihydrogen Production System"
Chem. Commun. 2014, 50, 8065-8068
S. C. Eady, T. Breault, L. Thompson, N. Lehnert
"Highly Functionalizable Penta-Coordinate Iron Hydrogen Production Catalysts with Low Overpotentials"
Dalton Trans. 2016, 45, 1138-1151
S. C. Eady, M. M. MacInnes, N. Lehnert
"A Smorgasbord of Carbon: Electrochemical Analysis of Cobalt-Bis(benzenedithiolate) Complex Adsorption and Electrocatalytic Activity on Diverse Graphitic Supports"
ACS Appl. Mater. Interfaces 2016, 8, 23624-23634
Erratum: ACS Appl. Mater. Interfaces 2017, 9, 15898-15899
S. C. Eady, M. M. MacInnes, N. Lehnert
"Immobilized Cobalt-Bis(Benzenedithiolate) Complexes: Exceptionally Active Heterogeneous Electrocatalysts for Dihydrogen Production from Mildly Acidic Aqueous Solutions"
Inorg. Chem. 2017, 56, 11654-11667
M. M. MacInnes, S. Hlynchuk, S. Acharya, N. Lehnert, S. Maldonado
"Reduction of Graphene Oxide Thin Films by Cobaltocene and Decamethylcobaltocene"
ACS Appl. Mater. Interfaces 2017, 9, 2004-2015
Literature:
[1] Rakowski DuBois, M.; DuBois, D. L. Acc. Chem. Res. 2009, 42, 1974-1982
[2] Nocera, D. G. Acc. Chem. Res. 2012, 45, 767-776
[3] Kim, D.; Sakimoto, K. K.; Hong, D.; Yang, P. Angew. Chem. Int. Ed. 2015, 54, 3259�3266
[4] Artero, V.; Chavarot-Kerlidou, M.; Fontecave, M. Angew. Chem. Int. Ed. 2011, 50, 7238-7266
[5] Stolley, R. M.; Helm, M. L. Nat. Chem. 2014, 6, 949-950
[6] Saveant, J.-M. Chem. Rev. 2008, 108, 2348-2378
[7] Walter, M. G.; Warren, E. L.; McKone, J. R.; Boettcher, S. W.; Mi, Q.; Santori, E. A.; Lewis, N. S. Chem. Rev. 2010, 110, 6446�6473
[8] Sala, X.; Romero, I.; Rodriguez, M.; Escriche, L.; Llobet, A. Angew. Chem. Int. Ed. 2009, 48, 2842-2852
|
