Our research
Chemical Dynamics: therory, computation and application

Modern computational chemistry strives to provide an atomistically detailed dynamical description of fundamental chemical processes. The strategy for reaching this goal generally follows a two-step program. In the first step, electronic structure calculations are used to obtain the force fields that the nuclei are subject to. In the second step, molecular dynamics simulations are used to describe the motion of the nuclei. The first step is always based on quantum mechanics, in the light of the pronounced quantum nature of the electrons. However, the second step is most often based on classical mechanics. Indeed, classical molecular dynamics simulations are rroutinely used nowadays for describing the dynamics of complex chemical systems that involve tens of thousands of atoms. However, there are many important situations where classical mechanics cannot be used for describing the dynamics. Our research targets the most chemically relevant examples of such processes.
1. Vibrational end electronic relaxation
The pathways of intramolecular energy redistribution within molecules and intermolecular energy transfer between molecules, which dictate chemical reactivity, are governed by the rates of these processes. The pronounced quantum nature of these processes is attributed to the large gap between vibrational and electronic energy levels.
2. Proton and electron transfer reactions
The elementary steps of many complex chemical processes are based on such reactions. Their pronounced quantum nature is attributed to the light mass of protons and electrons, which often give rise to quantum tunneling and zero-point energy effects.
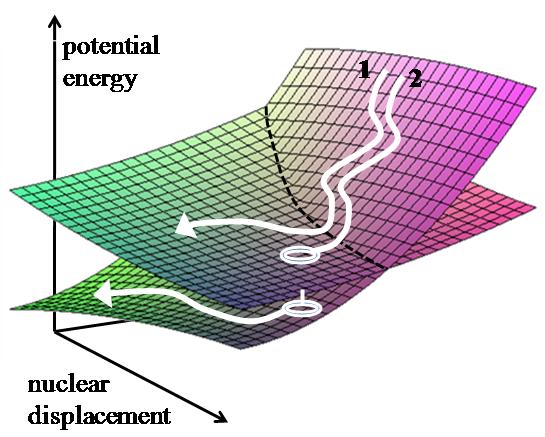
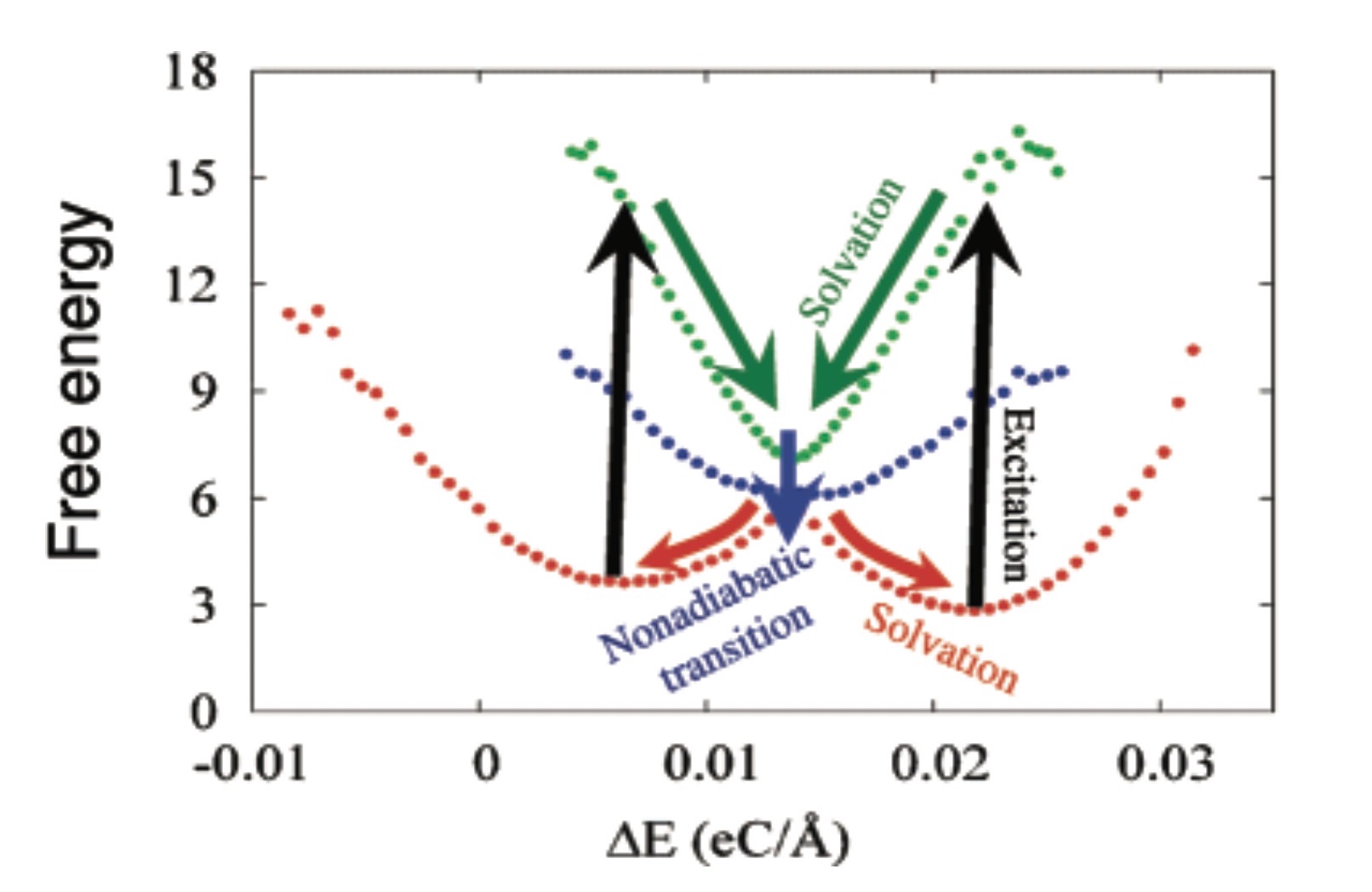
3. Nonadiabatic dynamics
Such dynamics underlie photochemistry and nonlinear spectroscopy is quantum in nature since it involves simultaneous motion on several potential surfaces that correspond to different electronic or vibrational states. The inherently quantum nature of the underlying molecular dynamics makes simulating such processes in complex molecular systems challenging.
Despite many advances in quantum-mechanically exact simulation techniques for general anharmonic Hamiltonians, semiclassical and mixed quantum-classical approaches remain the only viable methodologies for large-scale simulations of complex technologically and biologically relevant molecular systems. Such methods include Fewest-Switches Surface-Hopping (FSSH), the Ehrenfest method, partially linearized path integral approaches, Trotter-Based Surface-Hopping (TBSH), the Forward–Backward Trajectory Solution (FBTS) of the quantum-classical Liouville equation (QCLE), the Linearized Semiclassical (LSC) method, the Poisson Bracket Mapping Equation (PBME), and the symmetrical Quasi-Classical (SQC) method.

4. Chemical Dynamics in Cavity
Recent experimental realizations of strong coupling between optical cavity modes and molecular matter placed inside the cavity have opened exciting new routes for controlling chemical processes. Simulating the cavity-modified dynamics of complex chemical systems calls for the development of accurate, flexible, and cost-effective approximate numerical methods that scale favorably with system size and complexity.
The challenge involved in simulating the quantum molecular dynamics of such systems has to do with the fact that the computational effort involved in solving the time-dependent Schrodinger equation is exponentially larger than that involved in Newton's equations. As a result, a numerically exact solution of the Schrondinger equation is not feasible for a system that consists of more than a few atoms. The main research thrust of the Geva group is aimed at developing rigorous and accurate mixed quantum-classical, quasi-classical and semiclassical methods that would make it possible to simulate equilibrium and nonequilibrium quantum dynamics of systems that consist of hundreds of atoms and molecules. We put emphasis on applications to experimentally-relevant disordered complex condensed phase systems such as molecular liquids, which serve as hosts for many important chemical processes. We also specialize in modeling and analyzing different types of time resolved electronic and vibrational spectra that are used to probe molecular dynamics in those systems, often in collaboration with experimental groups.