|

|
Modeling Flavodiiron Nitric Oxide Reductases (FNORs)
Flavodiiron Nitric Oxide Reductases (FNORs) belong to the class of flavodiiron proteins (FDPs) and are important enzymes in bacterial pathogenesis [1]. FDPs were
first recocknized as dioxygen scavengers, protecting microanaerobs from oxidative stress by reducing residual O2 to H2O. Later, Gardner realized that somw FDPs were
specifically expressed as a response to NO, reducing it to less toxic N2O [2]. These FNORs allow these microbes to fight off nitric oxide produced by our macrophages for immune defense [3].
In this way, FNORs facilitate bacterial infections of mammalian organisms.
FNORs are therefore of significant interest in bacterial pathogenesis, and evaluating the mechanism of these enzymes contributes to the greater goal of finding new ways to fight bacterial infection.
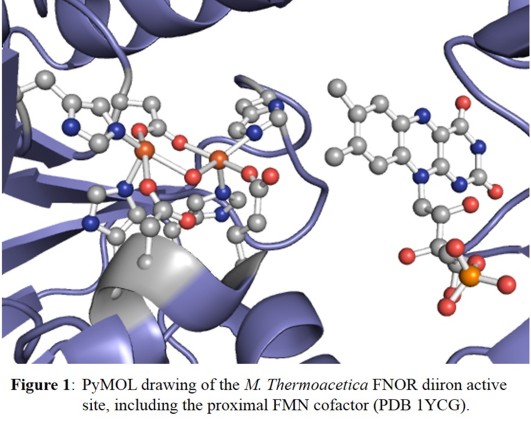
Crystallographic studies on Morella thermoacetica and Thermotoga maritima FDPs reveal an active site consisting of a non-heme diiron core, with two central iron atoms, each coordinated
by two histidine and one carboxylate ligands [4]. In addition, the two iron centers are bridged by water/hydroxide and a carboxylate ligand, as shown in Figure 1.
In either case, each iron is five-coordinate with an open 6th coordination site for substrate binding. Additionally, a flavin (FMN) cofactor rests across the subunit interface, within 4-6 Å of the
active site (see Figure 1) and is within range to facilitate rapid electron transfer.
Different mechanisms for NO activation and reduction have been proposed for FNORs (Scheme 1) [1]. Recent enzyme studies on the T. maritima FDP by Kurtz and coworkers have shown that a diferrous dinitrosyl
is formed as the critical intermediate preceding N2O generation [5]. In this case, NO sequentially binds the active site prior to the rate determining (N2O producing) step, implicating the
diferrous dinitrosyl complex, or [{FeNO}7]2 in the Enemark-Feltham notation, as the catalytically competent intermediate as indicated in Scheme 1.
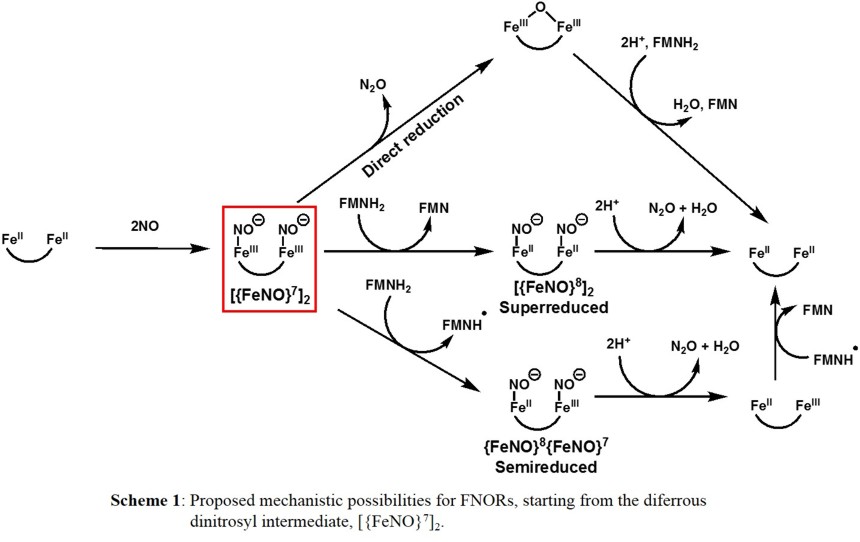
From the [{FeNO}7]2 intermediate formed
first, the two NO units may react in a direct N-N coupling pathway, where no external reductant is necessary, to facilitate N-N bond formation, generating N2O and the diferric form of the diiron active site.
The latter is subsequently re-reduced by FMNH2 to the diferrous active state of the enzyme. This pathway is supported by kinetic studies on the T. maritima FDP [5-6]. This finding is quite surprising, as mono- and
dinuclear non-heme {FeNO}7 model complexes have been found to be stable in solution, due to the rather covalent nature of the Fe-NO bond. Accordingly, these complexes have no propensity to form
N2O without the assistance of an external reductant. It is therefore unclear whether the direct NO reduction pathway accurately reflects the mechanism of NO reduction in designated FNORs under turnover.
For this reason, we have employed model complexes to further identify chemically facile pathways for efficient NO reduction by diiron active sites.
Diferrous dinitrosyl adducts have been prepared for ribonucleotide reductase (RNR) and soluble methane monooxygenase (sMMO) as models for corresponding O2 adducts, and one
corresponding model complex, [Fe2(Et-HPTB)(O2CPh)(NO)2](BF4)2, which shows end-on bound {FeNO}7 units [7]. Since the NO adducts of RNR and sMMO
only show negligible reactivity towards N2O formation (the model complex has no reactivity), their properties were not further studied.
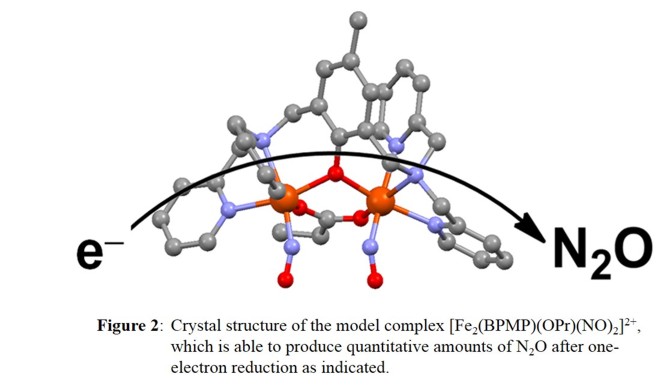
We prepared the diferrous dinitrosyl complex, [Fe2(BPMP)(OPr)(NO)2]X2 (X = BPh4-, OTf-, BF4-; OPr- = propionate), which is stable in solution and was structurally characterized (see Figure 2).
Each iron center has a pseudo-octahedral coordination environment. The BPMP- ligand induces a slight asymmetry in the complex, where one {FeNO}7 unit has the tertiary amine as the trans ligand to NO,
while the other one has a pyridine bound trans to NO. This complex exhibits antiferromagnetic coupling between the two S=3/2 {FeNO}7 units, as in FDPs and other diiron model complexes with short Fe-Fe bond
distances and several bridging ligands. Importantly, upon addition of one equivalent of reductant (cobaltocene, CoCp2), this complex shows fast and quantitative N2O generation (as indicated in Figure 2), which we call
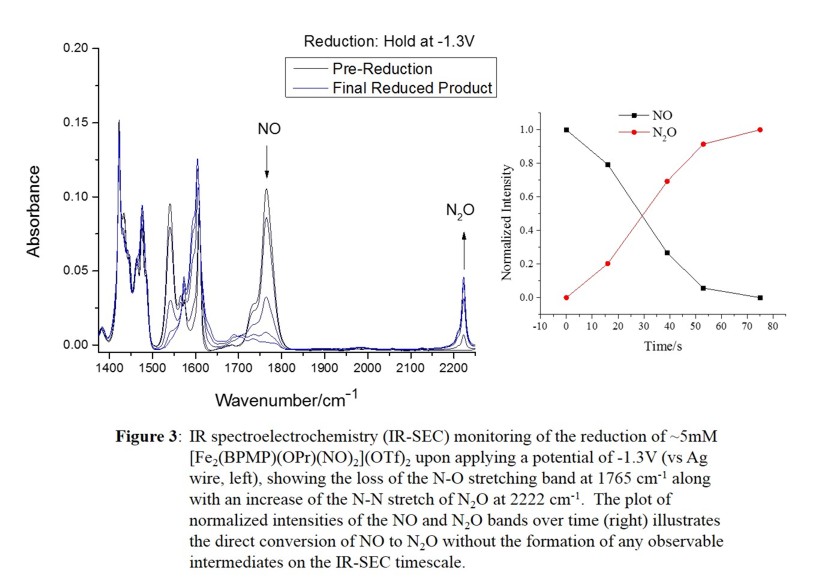
the semireduced mechanism. This is a new mechanistic possibility for FNORs that has previously not been considered. Our results show that this process is extremely facile, with an estimated activation energy of only a few kcal/mol
(based on stopped-flow IR results, spectro-electrochemistry as shown in Figure 3, and low-temperature studies). We further characterized the reaction product by UV-Vis, IR, Mössbauer, and EPR spectroscopy and cyclic voltammetry.
DFT calculations support these results, and provide a
mechanistic roadmap of how N-N bond formation and N2O generation take place.
These findings identify the semireduced pathway as a highly efficient means of NO reduction by a diiron core, implicating this as a mechanistic possibility for FNORs and synthetic electrocatalysts for NO degradation.
In addition, these results demonstrate that reduction is a very potent way to activate otherwise stable {FeNO}7 complexes to become more reactive.
Even though evidence for the direct NO reduction pathway has been presented for FDPs, no synthetic model complex has been able to mediate this reaction so far, and hence, the feasibility of this process is still in question.
Based on our results for [Fe2(BPMP)(OPr)(NO)2]X2 and the ability of this complex to mediate N2O formation via the semireduced pathway, we hypothesized that a complex with more reducing
iron centers might allow for the direct reduction of NO to N2O from the [{FeNO}7]2 intermediate. To test the feasibility of this idea, we prepared the model complex
[FeIII2((Py2PhO2)MP)(OPr)2](OTf) where two of the pyridine groups in the H-BPMP ligand used previously are replaced by more electron-donating phenolate groups. This complex was
further characterized through spectroscopic methods and electrochemical studies. Surprisingly, electrochemical studies show that the FeIIIFeIII/FeIIIFeII and
FeIIIFeII/FeIIFeII redox potentials are shifted more negatively by about 1 V in the presence of the two phenolates, as shown in Figure 4.
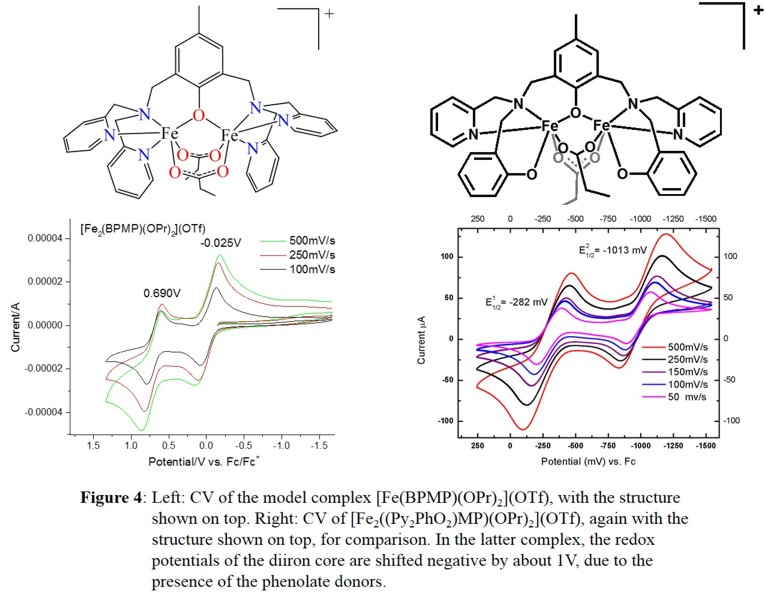
[FeIII2((Py2PhO2)MP)(OPr)2](OTf) can be reduced (using CoCp2) cleanly to the diferrous complex, which was structurally characterized. Excitingly, upon reaction with NO, this complex
produces quantitative amounts of N2O, without the need to add additional electrons to activate the {FeNO}7 units. Our results therefore provide direct support for the recent proposal that FNORs mediate NO detoxification
via direct NO reduction by their diferrous active sites, without the involvement of the flavin cofactor in the reaction. Here, proper tuning of the redox potential of the diiron complex is key in order to enable this reaction.
References:
T. C. Berto, A. Speelman, S. Zheng, N. Lehnert
"Mono- and Dinuclear Non-Heme Iron-Nitrosyl Complexes: Models for Key Intermediates in Bacterial Nitric Oxide Reductases"
Coord. Chem. Rev. 2013, 257, 244-259
S. Zheng, T. C. Berto, E. Dahl, M. B. Hoffman, A. L. Speelman, N. Lehnert
"The Functional Model Complex [Fe2(BPMP)(OPr)(NO)2](BPh4)2 provides Insight into the Mechanism of Flavodiiron NO Reductases"
J. Am. Chem. Soc. 2013, 135, 4902-4905
(highlighted in Chemical & Engineering News, April 1, 2013; see: View Article)
N. Kindermann, A. Schober, S, Demeshko, N. Lehnert, F. Meyer
"Reductive Transformations of a Pyrazolate-Based Bioinspired Diiron Dinitrosyl Complex"
Inorg. Chem. 2016, 55, 11538=11550
M. Jana, N. Pal, C. J. White, C. Kupper, F. Meyer, N. Lehnert, A. Majumdar
"A Functional Mononitrosyl Diiron(II) Complex Mediates the Reduction of NO to N2O with Relevance for Flavodiiron NO Reductases"
J. Am. Chem. Soc. 2017, 139, 14380-14383
C. J. White, A. L. Speelman, C. Kupper, S. Demeshko, F. Meyer, J. P. Shanahan, E. E. Alp, M. Hu, J. Zhao, N. Lehnert
"The Semireduced Mechanism for Nitric Oxide Reduction by Non-Heme Diiron Complexes that Model Flavodiiron NO Reductases"
J. Am. Chem. Soc. 2018, 140, 2562-2574
C. Van Stappen, N. Lehnert
"Mechanism of N-N Bond Formation by Transition Metal-Nitrosyl Complexes: Modeling Flavodiiron Nitric Oxide Reductases"
Inorg. Chem. 2018, 57, 4252-4269
H. T. Dong, C. J. White, B. Zhang, C. Krebs, N. Lehnert
"Non-Heme Diiron Model Complexes Can Mediate Direct NO Reduction: Mechanistic Insight into Flavodiiron NO Reductases"
J. Am. Chem. Soc. 2018, 140, 13429-13440
N. Lehnert, K. Fujisawa, S. Camarena, H. T. Dong, C. J. White
"Activation of Non-Heme Iron-Nitrosyl Complexes: Turning up the Heat"
ACS Catal. 2019, 9, 10499-10518
M. Jana, C. J. White, N. Pal, S. Demeshko, F. Meyer, N. Lehnert, A. Majumdar
"Functional Models for the Mono- and Dinitrosyl Intermediates of FNORs: Semireduction
versus Superreduction of NO"
J. Am. Chem. Soc. 2020, 142, 6600-6616
Literature:
[1] Kurtz, D. M., Jr. Dalton Trans. 2007, 35, 4115-4121.
[2] Gardner, A. M.; Helmick, R. A.; Gardner, P. R. J. Biol. Chem. 2002, 277, 8172-8177.
[3] Khatua, S.; Majumdar, A. J. Inorg. Biochem. 2015, 142, 145-153.
[4] Silaghi-Dumitrescu, R.; Kurtz, D. M.; Ljungdahl, L. G.; Lanzilotta, W. N. Biochemistry 2005, 44, 6492-6501.
[5] Caranto, J. D.; Weitz, A.; Hendrich, M. P.; Kurtz, D. M., Jr. J. Am. Chem. Soc. 2014, 136, 7981-7992.
[6] Caranto, J. D.; Weitz, A.; Giri, N.; Hendrich, M. P.; Kurtz, D. M., Jr. Biochemistry 2014, 53, 5631-5637.
[7] Feig, A. L.; Bautista, M. T.; Lippard, S. J. Inorg. Chem. 1996, 35, 6892-6898.
|
