|

|
Electronic Structure of Ferric Heme Nitrosyls
The electronic structure of ferric heme-nitrosyls (or {FeNO}6 complexes in the Enemark-Feltham notation)
with axial N-donor coordination is best described as a low-spin Fe(II)
center with a bound NO+ ligand, in agreement with the structural and spectroscopic properties of these complexes. These species are
charaterized by linear Fe-N-O units, short Fe-NO bond lengths, and N-O stretching frequencies around 1900 cm-1 [1], which strongly supports
this electronic structure description. The Fe(II)-NO+ ground state of ferric heme-nitrosyls is isoelectronic with Fe(II)-porphyrin CO complexes.
Correspondingly, the Fe-NO interaction in this state is dominated by two strong π-backbonds between Fe(II) and NO+,
which are in fact stronger than the backbonds in the corresponding CO complexes. Figure 1 shows contour plots of the resulting antibonding MOs (left and middle),
which have about 70% π* and 30% d contributions.
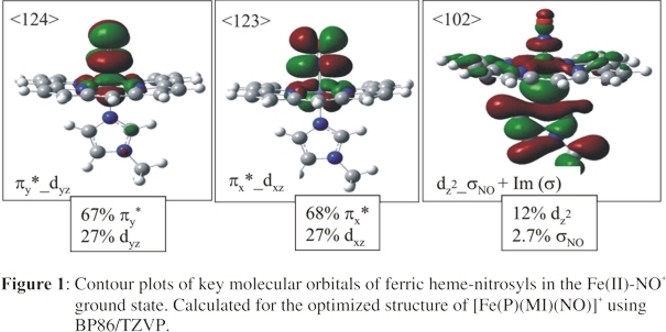
In contrast, the Fe-NO σ bond in these complexes is rather weak, as indicated by the corresponding antibonding MO in Figure 1, right,
which only has 3% NO contribution. The overall strength of the Fe-NO bond is reflected by the Fe-NO stretching frequencies in these complexes, which are typically
observed in the 580-600 cm-1 range [1,2].
These properties of the Fe-NO+ ground state of ferric heme-nitrosyls are consistent, but lead to one puzzeling question: if the Fe-NO bond is that strong
in the ground state of these complexes, then why is the binding constant of NO to ferric hemes so small, making these complexes susceptible to NO loss? This apparent
contradiction is evident when the properties of analogous ferrous and ferric heme nitrosyls are compared on the basis of either thermodynamic or spectroscopic criteria.
Experimental complex formation constants (Keq) of ferrous heme-nitrosyls are typically in the range of Keq = 1011 - 1012 M-1,
which translates to NO complex formation energies, ΔG0, of about -15 to -16 kcal/mol. In contrast, in ferric heme nitrosyls
Keq = 103 - 105 M-1, which corresponds to ΔG0 of -4 to -7 kcal/mol [3]. Therefore, the Fe-NO bond is
thermodynamically much stronger in the ferrous compared to the ferric case. In contrast, the Fe-NO stretching frequency and force constant is 580 cm-1 and
3.92 mdyn/Å for ferric [Fe(TPP)(MI)(NO)]+ (TPP2- = tetraphenylporphyrin dianion) compared to 437 cm-1 and 2.57 mdyn/Å for ferrous [Fe(TPP)(MI)(NO)]. This demonstrates that
spectroscopically, the Fe-NO bond is stronger in the ferric compared to the ferrous case, which contradicts the observed thermodynamic trend. Hence, NO is a weak
ligand to ferric heme (from thermodynamics), but at the same time, makes a strong Fe-NO bond (from spectroscopy)! This puzzeling observation can be explained when the
complete potential energy surface (PES) for the binding of NO to ferric hemes is considered.
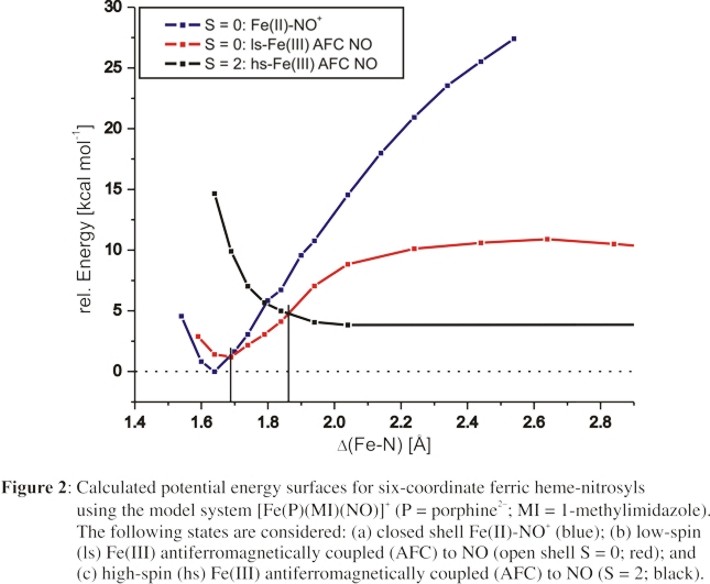
Figure 2 shows the potential energy surface derived for the Fe(II)-NO+ ground state (blue curve, S = 0) of ferric heme-nitrosyls plus energy surfaces of two
additional key states: the open shell singlet low-spin (ls) Fe(III)-NO(radical) alternative ground state, where low-spin (ls) Fe(III) is antiferromagnetically coupled to
NO (red curve, S = 0), and the corresponding high-spin (hs) state hs-Fe(III)-NO(radical) (black curve, S = 2), which corresponds to the product state upon dissociation of
NO (since the five-coordinate ferric heme-imidazole complex is high-spin). Figure 2 shows that the PES for the Fe(II)-NO+ ground state is very steep with an estimated
dissociation energy of >30 kcal/mol, which reflects the unfavorable dissociation of the complex into Fe(II) and NO+. Importantly, this explains the spectroscopically
observed strength of the Fe-NO bond in ferric heme-nitrosyls: this is in fact a property of the Fe(II)-NO+ ground state of these complexes.
Interestingly, the alternative ground state of ferric heme-nitrosyls, ls-Fe(III)-NO(radical) (S = 0), is surprisingly close in energy to the Fe(II)-NO+ ground state,
with a DFT-calculated energy difference of only ~1 kcal/mol. This small energy difference is clearly an underestimate, as evident from the experimental properties of these
complexes, which do not indicate the presence of a low-lying excited state with a distinctively weaker Fe-NO bond. Furthermore, the Fe(II)-NO+ ground state is
likely stabilized by configuration interaction (CI) via the doubly excited electronic state within the Fe-NO π-backbond. Taking this into consideration,
the energy surface of the Fe(II)-NO+ ground state crosses that of the ls-Fe(III)-NO(radical) state at an Fe-NO distance of only about 1.70 - 1.76 Å. This is
accompanied by the transfer of one electron from Fe(II) back to NO+, forming a ls Fe(III) center with a bound NO radical.
Upon a further elongation of the Fe-NO bond, the ls-Fe(III)-NO(radical) energy surface crosses that of the hs-Fe(III)-NO(radical) state (S = 2) at a Fe-NO distance of about 1.9 Å
(see Figure 2). This transition corresponds to a spin crossover of the iron(III) center, and is related to the fact that the five-coordinate ferric heme product is actually in the high-spin state.
Importantly, the hs-Fe(III)-NO(radical) energy surface is dissociative with respect to the Fe-NO bond. This causes a dramatic drop in the thermodynamic stability of the ferric Fe-NO complex,
from about -10 kcal/mol in the ls-Fe(III)-NO(radical) state to only about -4 kcal/mol (calculated) in the hs-Fe(III)-NO(radical) state. Therefore, the properties of the hs-Fe(III)-NO(radical)
energy surface determine the thermodynamic weakness of the Fe-NO bond in ferric heme-nitrosyls, and the large dissociation rate constant of NO. In other words, once the system is in the
hs-Fe(III)-NO(radical) electronic state, the dissociative nature of the corresponding energy surface will actually drive the NO away from the metal center, causing a distinct increase
in the NO dissociation rate compared to ferrous heme-nitrosyls. Hence, the experimentally derived Fe-NO force constant is not a measure for the stability of the Fe-NO bond in this case.
These quantities are actually completely unrelated, because they depend on the properties of different electronic states. In this way, NO can form a strong Fe-NO bond and at the same time,
be a weak ligand to a ferric heme.
More recently, we devised a new method for the clean bulk synthesis of ferric heme-nitrosyl complexes in high purity. This method is based on the
chemical or electrochemical oxidation of corresponding {FeNO}7 precursors. We used this method to obtain the five- and six-coordinate complexes [Fe(TPP)(NO)]+
and [Fe(TPP)(NO)(MI)]+ (TPP = tetraphenylporphyrin dianion; MI = 1-methylimidazole), and demonstrate that these complexes are stable
in solution in the absence of
excess NO gas. This is in stark contrast to the often cited instability of such {FeNO}6 model complexes in the literature, which we attribute to the common presence of halide impurities.
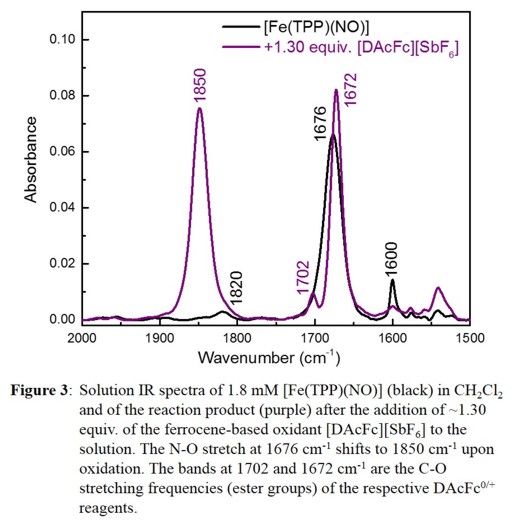
This is avoided in our new approach for the synthesis of {FeNO}6 complexes via oxidation of pure {FeNO}7 precursors. Figure 3 shows the chemical oxidation
of [Fe(TPP)(NO)] to [Fe(TPP)(NO)]+ as example, followed by IR spectroscopy. Based on these results, {FeNO}6 complexes in proteins do NOT show an increased stability compared to
model complexes towards NO loss. We also prepared the halide-coordinated complexes [Fe(TPP)(NO)(X)] (X = Cl-, Br-), which correspond to the elusive, key reactive intermediate
in the so-called autoreduction reaction, which is frequently used to prepare {FeNO}7 complexes from ferric precursors.
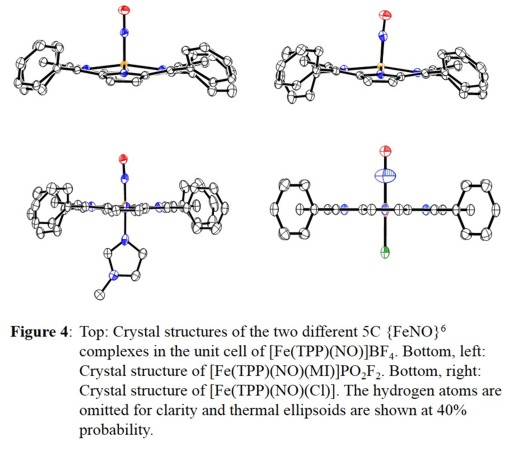
All complexes were characterized using X-ray crystallography, as shown in Figure 4, and also by different spectroscopic methods. Based on the vibrational data (IR and NRVS), further insight into
the electronic structure of these {FeNO}6 complexes, in particular with respect to the role of the axial ligand trans to NO, is obtained. Interestingly, for six-coordinate {FeNO}6
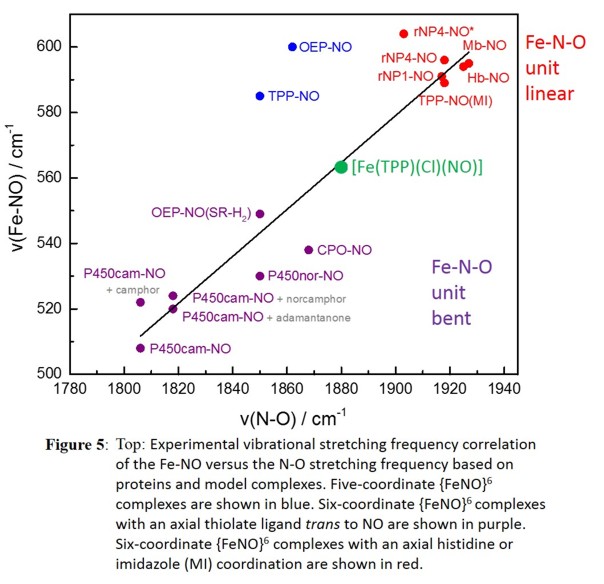
complexes with anionic axial ligands, a correlation of the ligand donor strength and the Fe-N-O bond angle has been derived [1]. Whereas complexes with axial, neutral N-donor ligands show linear
Fe-N-O units (see Figure 4), the presence of an anionic thiolate donor, for example in the model complex [Fe(OEP)(NO)(SR-H2)] (where SR-H2 = S-2,6-(CF3CONH)2C6H3),
leads to a bending of the Fe-N-O moiety with an Fe-N-O angle of about 160o [4]. In addition, the axial ligand donor strength is also correlated with N-O and Fe-NO stretching frequencies as
depicted in Figure 5. Interestingly, [Fe(TPP)(NO)(Cl)] is unique and falls in the region between the neutral N-donor and thiolate-ligated complexes on the graph. This would indicate the Cl-
ligand is a moderate donor, but does not explain the fact that the Fe-N-O unit is completely linear (see Figure 4). Therefore, the Cl- ligand is a moderate σ- and
isotropic π-donor that causes a simultaneous weakening of the Fe-NO and N-O bonds, evidenced by vibrational spectroscopy, but the isotropic π-donating
ability of Cl- seems to cause the linearity of the Fe-N-O unit unlike other anisotropic π-donor ligands. This emphasizes the importance of (vibrational) spectroscopy
over structural data for the characterization of metal-ligand bond strength. More work is required to fully understand the electronic reasons for Fe-N-O bending in thiolate-coordinated ferric heme-NO complexes
and the significance of this effect for NO activation in Cyt. P450s (especially Cyt. P450 NO reductase) [5].
References:
V. K. K. Praneeth, F. Paulat, T. C. Berto, S. DeBeer George, C. Näther, C. D. Sulok, N. Lehnert
"Electronic Structure of Six-Coordinate Iron(III)-Porphyrin NO Adducts: the Elusive Fe(III)-NO(radical) State
and Its Influence on the Properties of these Complexes"
J. Am. Chem. Soc. 2008, 130, 15288-15303
N. Lehnert, W. R. Scheidt, M. W. Wolf
"Structure and Bonding in Heme-Nitrosyl Complexes and Implications for Biology"
Struct. & Bonding 2014, 154, 155-224
(special issue: Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine II)
A. B. McQuarters, J. Kampf, E. E. Alp, M. Hu, J. Zhao, N. Lehnert
"Ferric Heme-Nitrosyl Complexes: Kinetically Robust or Unstable Intermediates?"
Inorg. Chem. 2017, 56, 10513-10528
(Editors' Choice)
D. J. Thomas, N. Lehnert
"The Biocoordination Chemistry of Nitric Oxide with Heme and Non-Heme Iron Centers"
in: 'Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering'; Reedijk, J., Ed.,
Elsevier, 2017,
doi: 10.1016/B978-0-12-409547-2.11678-6
Literature:
[1] Goodrich, L. E.; Paulat, F.; Praneeth, V. K. K.; Lehnert, N. Inorg. Chem. 2010, 49, 6293-6316.
[2] Soldatova, A. V.; Ibrahim, M.; Olson, J. S.; Czernuszewicz, R. S.; Spiro, T. G. J. Am. Chem. Soc. 2010, 132, 4614-4625.
[3] Lehnert, N.; Berto, T. C.; Galinato, M. G. I.; Goodrich, L. E., The Role of Heme-Nitrosyls in the Biosynthesis, Transport, Sensing, and Detoxification of
Nitric Oxide (NO) in Biological Systems: Enzymes and Model Complexes, in: K.M. Kadish, K.M. Smith, R. Guilard (Eds.) The Handbook of Porphyrin Science,
World Scientific, New Jersey, 2011, Vol. 14, pp. 1.
[4] Xu, N.; Powell, D. R.; Cheng, L.; Richter-Addo, G. B. Chem. Commun. 2006, 2030-2032.
[5] McQuarters, A. B.; Wirgau, N. E.; Lehnert, N. Curr. Op. Chem. Biol. 2014, 19, 82-89.
|
